The Hartree-Fock method
Oktober - November 1995 version
As mentioned in the introduction,
the Hartree-Fock method is based on the
independent particle approximation. It means that each electron
is assumed to move in an
average potential created by the nucleus and the other electrons.
The approximative Hamiltonian can then be written as a
sum of one-particle Hamiltonians
(1) 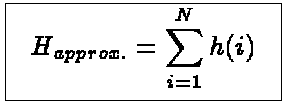
where N is the number of electrons in our system. The simplest form of an
eigenfunction to Happrox. will be a product function
(2) 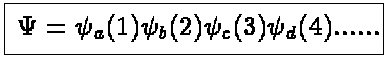
where a , b etc stand for the quantum numbers
necessary to specify a
single electron state. Any permutation of the single-particle functions,
which we can call spin-orbitals, will
also lead to an eigenfunction of Happrox. .
However, in order to satisfy the Pauli exclusion principle the total wave
function, Psi, must be antisymmetric with respect to the interchange of any
two of the electrons. We can form a wave function with this property by
antisymmetrizing the product function above. We write this
antisymmetrized wave function as
(3) 
and it is called a Slater determinant.
As a simple example consider an atom with two electrons, than
the antisymmetrized wave function will be
(4) 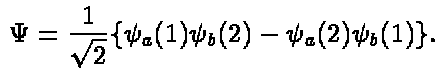
If we want the total wave function, Psi to consist of
one Slater determinant,
how do we chose the ``best'' one?
The expectation value of the
total energy for a state represented by the Slater determinant
is given by the expectation value of the total Hamiltonian
(5) 
The ``best'' determinant for the ground state should be the one which
minimize the expectation value of H.
We thus minimize the energy with respect
to changes in the Slater determinant
Psi.
This leads to the
Hartree-Fock equations
Clicking on the formula will bring you it's latex source